

過去数十年のキャリアにわたって、私は薬物の安全性を研究することでアメリカ国民を守るために数えきれないほどの時間を費やしてきました。私の教育とキャリアは、約 XNUMX の大学、大手製薬会社、そして XNUMX つの大統領政権下での FDA を経てきました。医薬品の安全性では、ある人が医薬品を服用しても有害事象がゼロであるのに、別の人が同じ製品を服用しても、永久的な障害や死亡に至るまでの副作用が起こる可能性がある理由を考慮します。デフォルトでは、医薬品の安全性の研究では、製造および医薬品の品質の非臨床面も考慮されます。
医薬品の品質は医薬品の安全性を評価する上で不可欠な要素であるため、アメリカ人を保護するという私の取り組みが、世界初の「」の概念化と創設につながりました。分析薬局」は、インドや中国などからの医薬品を患者に配布する前に科学的に検証することを使命としています。残念ながら、倫理や患者保護を優先するあまり、その会社の財務管理は、 広範囲 FDA違反 そして裁判官から次の行為をしたとして告発されている 誤った科学的主張 (これらはすべて、私が退職した後に偶然に起こりました)。
医薬品の品質を外部から確認することなしに、アメリカ人は製品の純度の評価と確認を FDA と製造業者に完全に依存しています。新型コロナウイルスのmRNA注射に関しては、医薬品の安全性が注目すべき問題であることが示されている。残念ながら、mRNA 注入について独自の分析を行おうとする人がいれば、 比較するための適切に詳細な成分リストがなく、純度を適切にテストする方法に関する確立された規制手法にもアクセスできません。.
メーカーだからね & FDA は、mRNA の配列と脂質ナノ粒子 (LNP) の特性 (半減期、LNP 構造、表面修飾、用量あたりの LNP の数/種類、および結合点など) を含む、これらの mRNA 注射剤のすべての成分を考慮します。 mRNA 鎖は特定されない、または「企業秘密」となります。
それに加えて、FDA はさらに次のことを考慮しています。 方法論 mRNA注入の純度をテストする方法も企業秘密です。
超党派の支援と数千億ドルの納税者がいるが、透明性はない?
トランプ政権とバイデン政権の両方が、Covid mRNAの知的財産権を解除するという点まで、mRNA注射に関する完全な透明性を提案していたにもかかわらず、Covid mRNAの秘密は存在する。それにもかかわらず、FDAと製造業者は両方とも、これらのショットに関する基本データを含む特許を企業秘密として許可し、厳重に管理している。すべての新型コロナウイルスワクチンメーカーがワクチン接種を受けているにもかかわらず、彼らはそうしている。 数億ドルの納税者 による フォーブス/スタティスタ 出版物。
医薬品の安全性疫学を研究することは十分に困難です。検証可能な製品の純度/一貫性がなければ、完全な安全性評価は不可能です。
すべての成分と品質管理措置の完全な透明性が重要なのは、それらが数億ドルという多額の税金で賄われているからだけでなく、新型コロナウイルスmRNA注射の安全性と有効性について多くの疑問が生じているためである。
非常に複雑であることに加えて、その承認は規制当局によって迅速に行われました。 XNUMX年未満。ほとんどの薬やワクチンは通常、 10年 安全性/有効性を十分にテストし、審査して承認します。成分が完全に新規で非常に複雑であり、大規模に投与される種類としては初めてであることに加えて、次のような開発が行われました。 長期的な臨床安全性/毒性評価と疫学調査が急がれたが、発売前に完全には解明されていない可能性が高い。
FDA の成分検証、透明性、「真実性」に関する前例は 1800 年代にまで遡ります。
分析検証と成分の透明性、またはボトルの中身が表示されている「ラベル表示の真実」 の提出が必要です 記載されている成分と一致するように FDA の設立よりも前の、1862 年に遡ります。。今日の FDA は、実際には米国農務省に雇用された XNUMX 人の「化学科」職員から始まりました。
粗悪品、(変質または有毒成分) 誤ったブランド名 (虚偽のラベルが含まれているか、誤解を招くものであるか、誤った医療上の主張が含まれている)、または 誤表示 (製品ラベルに記載されていない成分が含まれている)はすべて、アメリカで長く醜い歴史を持っています。成分不正を分析し検出するための技術的プロセスが開発されたのは 19 年になってからであり、その悪質さは 1862 世紀初頭から中期にピークに達したと考えられていました。あるいは、少なくともそれが識別可能になったのはその頃でした。それ以前は、自らを「医師」と呼ぶ、いわゆる「旅する医療マン」が(常に疑わしい、または存在しない資格を持って)「万能薬」製品のボトルを売り歩いていましたが、その成分ラベルには、次のような漠然とした、または無害な内容しか記載されていませんでした。 「ビタミン""ハーブエキス、」または「ヘビ油」 – または、成分リストがまったくないこともよくあります。
当時、多くの敬虔な清教徒のニューイングランド人は、宗教上の理由から、 決して アルコールに触れる人は、これらの行商人からこれらの溶液を購入し、知らず知らずのうちにだまされて、アルコールだけでなく、アヘンやコカインなどの麻薬を含む溶液を消費することになります。ばかばかしいほど広範な病気の宝庫を改善するという口実の下で、患者は代わりに罰的な依存症を発症したり、初期の「麻薬売人」によって健康に悪影響を受けたりしました。
問題が大きくなるにつれて、連邦政府も注目し始めました。最終的には、 純粋な食品医薬品法 1906 年に可決され、食品医薬品局 (FDA) の創設につながりました。
[FDA は 形成的 医薬品に真実のラベル表示があり、純度と強度に関する一定の基準を満たしていることを保証する義務。
120年近い歴史があることを忘れないでください。 真実の表示要件 mRNA 検証試験と成分の透明性について読み進めながら、1906 年の純粋食品医薬品法の「純度」の部分について説明します。]
FDA規制の製品に対してどのような「真実」で「純粋な」成分検証試験が行われていますか?
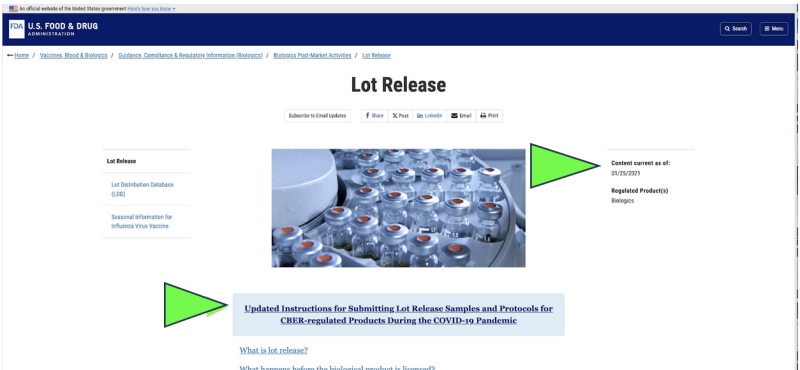
2021 年に遡ると、FDA は米国の医薬品の品質の監視を開始することを選択しました。 リモートコレクション of サンプルの郵送提出 新型コロナウイルスのパンデミックによる施設の実際の検査の代替としての薬物の検査。それは合法でしたか?それが科学的に適切であると考えられるでしょうか?現在、パンデミックは終息したにもかかわらず、現在行われている唯一の公式医薬品放出試験は、 どれか 新型コロナウイルスmRNA医薬品 登場する 〜へ まだ メーカー提供の方法を介して FDA によって行われます。郵送されました」 によるサンプル 現在の FDA Web サイトのスクリーンショット。明らかに、「郵送」サンプリング方法は、直接の対面収集方法によるサンプルの直接収集とは大きく異なり、信頼性が低い可能性があります。それにもかかわらず、FDA は次のように主張しています。世界中で最高水準のサンプリングとテストに設立された地域オフィスに加えて、さらにローカルカスタマーサポートを提供できるようになります。」
さらに、FDA は、「郵送による」遠隔検査政策をさらに推進することを提案しています。 新たに提案されたガイダンス文書.
これは FDA 文書の「草案」としてのみ存在しますが、FDA の公式 Web サイトには次のことが示されています。 サンプルの郵送は少なくとも 2021 年 XNUMX 月からすでに実施されているようです。 FDAは、これらの郵送検査の結果を独自の検証として主張しているようだ。
さらに、 FDA草案の最初のページの下部 文書では「遠隔検査」の拡大を提案している。現在リストされているのは、 あらゆる FDAのFDA製品規制部門は、これが全庁的な政策提案であることを示唆している。
完全なリストには次のものが含まれます。
- 規制問題局
- 食糧政策対応局
- コンビネーション製品のオフィス
- 生物製剤評価研究センター
- 薬物評価研究センター
- デバイスおよび放射線健康センター
- 食品安全・応用栄養センター
- タバコ製品センター
- 獣医学センター
FDA による品質管理サンプリングの「郵送」は適切ですか?もし各州の保健省のレストラン検査が FDA の方針を反映していたらどうなるでしょうか?
この「郵送」サンプリング方法論も同様に不合理である。たとえば、州の保健局がレストランを監視する場合、保健局が潜在的な食品を検査できるように、メニューのさまざまな品目を検査施設に定期的に「郵送」するよう求めている。 - 生まれながらの汚染、および/またはレストランにメニュー項目自体をテストすることを約束するよう求める。もしそのレストランが中国だったらどうなるでしょうか?もしそのレストランがインドにあったとしたら?または、 詐欺と品質管理のひどい歴史 問題?
この方法論は、レストランと製薬会社の両方にとって受け入れがたいものです。その理由には、明らかな理由が含まれます。メーカーは、必ずしも代表的なバッチ サンプルではなく、希望するサンプルを送信できるということです。これは明らかに、FDA 検査官が施設全体の抜き打ち検査中にサンプルを入手するのと同じではありません。
レストランに例えると、もちろんすべてのレストランは 「A」グレードのサンプルを提出する それは必ずしも消費者が受け取るものを表しているわけではありません。

品質管理: 医薬品の「放出試験」とは何ですか?なぜ重要ですか?
現在、FDA はその品質と内容を監督しています。 $2.7 1兆 年間の製品価値、しかし重要な成分検証の評価と結果を抑制しているようです。 FDAは以下のことを行うことでアメリカ人を保護することになっている 包括的な 成分の正確性を保証するためのチェックサムとしての分析テスト。その結果は、資金を提供している納税者にとって透明であるべきである。 FDAの6.6億ドル 予算。その科学的検証を「薬学」といいます。リリーステスト」リリーステストは、リリーステストに使用されるさまざまな機器分析を含むプロセスを指す専門用語です。 包括的 製品の純度、濃度、一貫性、同一性、およびあらゆる種類の不純物を検査します。
FDA 全体は、1862 年からのあの XNUMX 人の「化学科」職員と、成分の透明性と検証の必要性から生まれました。現在、その従業員は急増し、 1,300 人の科学者とサポートスタッフからなる FDA 部門全体 医薬品放出試験による成分検証に専念していると推定されています。 FDA の 医薬品品質管理室 (OPQ) は、品質/不純物 (定性) または含有量 (定性) のばらつきがなく、医薬品が記載されている成分の含有量と正確に一致していることを確認することを目的としています。それを必要とするルールは非常に具体的かつ詳細に規定されています。 21 CFR§201.10.
FDA が品質管理のために mRNA 注射を検証する方法:
mRNA 注入によるテストの品質管理結果は、大規模で複雑であり、迅速に作成されるため、特に重要でした。納税者は mRNA 注入の品質を検証し、結果を共有するために FDA に依存していますが、FDA は と思われる mRNAの新型コロナウイルス製品に関する最も基本的な透明性さえ犠牲にして、メーカーの成分を保護する義務を負っている。 FDA はサンプルを収集しているようですが、その「郵送」方法には根本的に欠陥があります。さらに、FDA は、私が見つけたどこにもそれらの検査結果を共有していません。
言い換えれば、パンデミックの最中に、広範に導入された真新しい mRNA ショットが「ワープスピード」でアメリカ国民に押しつけられ、アメリカが FDA の品質/規制義務に最も依存していたとき、FDA は自己提出の「郵送」を受け入れていたのである。 「」の品質管理テストおよび/または結果。 FDAはそれを考慮しなかったのか mRNAメーカーは、製造への対応に「苦労しており[d]」、追いつくために「スクランブルをかけている」ことを認めた 製造プロセスで?さらに、mRNA成分のメーカーは、ニーズを満たすための取り組みは「前例のないもの」であると述べた。
このような声明は、消費者に品質に対する信頼を与えるものではなく、これらの複雑な製品の大幅な大型化を例示するものであり、当然のことながら、 特に警戒している パンデミックの有無にかかわらず、FDA による施設および製造製品の直接検査。例えば、ある mRNA 成分メーカーは、 50折り.
その新技術が「ワープスピード」で急いで押し進められている最中に、FDAの1,300人のOPQ科学者は誰もライブ検査を要求しなかったし、少なくとも潜在的に疑わしい「郵送」サンプルを要求すること以外のことをしようと申し出なかった。テスト用?
明らかな質問は次のとおりです。 なぜFDAはサンプルを直接収集しなかったのか?パンデミックが発生していても、FDAは防護服を着て施設を検査したり、あるいは現場で検査したりすることもできたはずだ。 非常に 少なくとも、薬局、病院、または販売店の倉庫からサンプルを収集することを選択しました。
mRNA 注射の成分をテストするための隠された方法論:
検査結果の欠如や「郵送された」疑わしいサンプリング結果を超えて、FDA は さらに 検証済みの方法論を隠蔽することで、他社が mRNA 注入の品質/純度について独自の独立した分析を行うことができなくなります。
成分リストと比較して薬物の純度と潜在的な汚染を独立して分析することは、世界初の製品を構想したときに私自身が試みたことです。 分析薬局。ただし、mRNA ショットは成分リストが完全に透明とは言えない新しい技術であるため、採用する必要がある検査方法は他の低分子薬の場合のように単純ではありません。保存、安定性、特異性、化学、感度、あるいは検証や結果を試験するための基本的な方法論さえ調べようとする者は、ばかばかしいほど侵襲的な編集を含む FDA 報告書によって阻止され、潜在的な評価方法についての最も基本的な科学的理解すらできなくなる。結果やテストの実施が不可能になる可能性があります。
心を痛める視覚的な例として、より長い FDA 規制概要 (以下に示す) の編集された XNUMX ページは、 127ページのドキュメント (そのうち 63 ページのみが共有されており、そのうち約 63% が編集されています) mRNA注入の純度、濃度、その他の分析尺度を評価する方法について。
それらの FDA (b)(4) 編集 「」に使用される指定された詳細な編集営業秘密および機密の商業情報または財務情報を保護する」しかし、研究/開発/製品に資金が提供されている場合、それを「商業的」とラベル付けるのは本当に適切でしょうか? 数億ドルの納税者?

成分リストや試験方法がなければ、FDA や製造業者以外の誰もが製品を確認する方法を正確に知ることは不可能です。 混入 (変質または有毒な成分) または 誤表示 (ヌクレオチド配列と 脂質ナノ粒子の構成 製品ラベルでは特に曖昧です)。
独立した方法論を使用した新しい予備データが証拠を示しているため、方法論の欠如は特に問題です。 mRNA の DNA 汚染 Covid 注射.
したがって、外部の個人が、mRNA ショットを検査して不純物を発見したと主張し、FDA または製造業者に回答を求めた場合、次のような返答が返されるでしょう。
- 結論を出すために検証済みの適切なテスト方法を使用していないため、分析は無効です。
そのために、独立した検査機関は、FDA が承認した文書 (つまり、 フィギュア 5) 次のように尋ねます。「わかりました。承認された方法論を使用してテストしたいと思います。それが何なのか教えていただけますか?」
- FDA や製造業者は次のような返答をするでしょう。採用された方法論に関して当社が開示したいもののうち、機密ではないものは、オンラインまたは FDA の FOIA リクエストを通じて見つけることができます。” …彼らが出会う場所 以下の大幅に編集された文書、ここで、遠隔的に意味のあるものはすべて (b)(4) 編集で覆われています。
行間を読むと、メーカーもアメリカの FDA も、自社以外の者に mRNA 注射の全成分を知られたくない、あるいは mRNA 注射の純度や一貫性をテストすることさえ望んでいないのは明らかです。
FDA 当局者によると、医薬品製造は 特定のユースケースに合わせることができ、運用インフラコストを削減することができる高可用性と効率性を備えた エラーが発生しやすい:
その他にもたくさんのグーグルの 医薬品の製造プロセスでは、問題が発生する可能性があり、実際に問題が発生します。 mRNA/LNP 注入による潜在的な不一致を超えて、定性的および定量的問題が関係しています。 あらゆる FDA 規制の医薬品。下院と上院でさえ、FDAがアメリカの医薬品サプライチェーンを確保できなかったという報告を正式に認めている。 大多数の アメリカの製薬会社 消費者エンドユーザー製品生産されています 海外ではインドや中国など、その他の低人件費国は、 高い品質管理に定評がある。連邦官報には次のような報告があふれています。 インドと中国の製造工場での違反.
FDA は、長い違反歴のある工場も含めて、FDA への「郵送」システムを通じてこれらの工場を認定しているのでしょうか?とんでもないことに、この質問に対する答えは、医薬品の品質に関心のある人なら非常に不快にさせるものです。
同時に シックスシグマ 自動車、コンピュータ、携帯電話、その他のハイテク製造においては、精度レベルは長い間、品質と安全性の目標となってきましたが、医薬品製造に関してはほとんど無視されてきたようです。
FDA 当局者は、医薬品製造における不正確性を 2 ~ 3σ (シグマ) と推定するデータを発表しました。 2σ品質は以下に相当します。 308,537 件の機会あたり 1,000,000 件の欠陥。 (医薬品製造に関しては、おそらく 1,000,000 万件をはるかに超えるミスが発生する可能性があります。)FDA は最高レベルのリーダーシップでこのことを認識しています。実際、現在 FDA医薬品品質局の責任者、マイケル・コプチャ氏 医薬品製造の不正確な性質を嘆き、上記のシックスシグマの計算さえ書いて出版しました。 バック2017で.
mRNA 産物および/またはその LNP の誤差の範囲は均等である可能性があります。 less ヌクレオチド材料や新規 LNP が含まれているため、2 ~ 3 σ よりも精度が高く (σ が低いほど、製品に誤りが多くなります)、開発、製造、発売が行われているにもかかわらず、低分子医薬品よりも大幅に複雑になります。ワープスピード。"
FDA とその当局者さえも製造上の固有の不正確性を認識しているため、 広いスポーツの世界でなぜ? FDAはmRNA技術の放出試験を資金提供しているアメリカ国民と公に共有することで安全性の使命を果たしていないのだろうか?
また 1862 年以前ですか? mRNA注射はアメリカ人が持っていない唯一の薬なのか 完全 成分情報?
mRNA ショットのシーケンス数やその他の重要な情報が明確ではないことは、FDA が承認した別の RNA ベースの薬とはまったく対照的です。 パティシラン(オンパトロ®)。 Onpattro は、公式 FDA 内で自社製品の配列、分子量、ミリグラム強度を透過的に提供します。 パッケージラベル 以下の抜粋に示すように:


Covid mRNA の欠如 用量特異性: 0.3mL (または 0.5mL) なにかの?
現時点では、新型コロナウイルス mRNA 注射剤の基本的な成分情報はまだありません。薬剤師は特定の情報を与えることしか知りません。 ボリューム どうやら何の疑問も持たずにそうしたようだった。通常、FDA の公式パッケージラベルには、その容量に含まれる実際の成分が詳しく記載されている必要がありますが、Covid mRNA ラベルについてはそうではありません。単に「剤形と強度」として 0.3 mL (または 0.5 mL) と記載されています。
さらに、高校生なら誰でもわかると思いますが、0.3/0.5mL は ボリュームではない 力。その 0.3/0.5 mL に何が含まれているかについては、次のような定量的な詳細はわかりません。たとえば、LNP 粒子は何個ですか?それらの LNP のサイズ/形態は何ですか?そのボリュームには mRNA 配列がいくつありますか?




これはFDAによって十分に透明性のあるもの、あるいは「真実の表示」として認められるものなのでしょうか?
上記の添付文書からの切り貼りの抜粋は、製造業者が用量に関して消費者と共有しているすべての情報です。これは、他のすべての FDA ラベルと比較してひどく不十分です。または、液体の量以上のことを知りたい人は誰でも30 または 100 mcg 濃度の不特定の mRNA 配列を注入します。
FDA が許可したこのラベルの驚くべき不正確さは、特に 120 年近く前のラベルと矛盾しているようです。食品や医薬品に真実のラベル表示を行い、純度と強度に関する一定の基準を満たすことを要求するに設立された地域オフィスに加えて、さらにローカルカスタマーサポートを提供できるようになります。」
これはFDAによって「真実の」成分リストとして認められるものなのでしょうか? (見る 21CFR §352, 21 CFR §201.10 「成分表示」および「不正ブランドの医薬品および機器」に関して。)
問題は、メーカー以外の誰も解読できない未知のまたは非特定の成分をリストすることは可能かということです。 本当に 「表示」の精神または法的要件を満たしているか?そのラベルはアメリカの FDA によって「真実」とみなされるものなのでしょうか? いずれにせよ、FDAはどちらの味方なのでしょうか。メーカーか消費者か?
直接指定されていないことに加えて、30 または 100 mcg 注射における LNP 鎖または mRNA 鎖の正確な数は推定することさえできません。 化学量論的に またはそれに基づいて アボガドロの数なぜなら、mRNA 配列、分子量、および/または LNP 成分/構成は、FDA の公式ラベルのどこにも記載されていないからです。
新型コロナウイルスのスパイクタンパク質をコードするmRNA鎖の数が、市中感染によって得られる新型コロナウイルスの接種量に比例するかどうか、どうやって知ることができるのでしょうか?答え: 彼らはできません.
新型コロナウイルスのmRNA注射です 適切なラベルが貼られている/間違ってラベルが貼られている?
21 CFR 211.125 「」を指定します医薬品のラベル表示業務で使用するために発行されるラベルは厳格に管理されなければなりません。」 しかし、FDAは、次の事実にもかかわらず、Covid mRNA注射の承認されたラベル表示に関して非常に緩かったようです。 mRNA ベースの Onpattro を含む他のすべての薬剤では、その情報が明記されています。歴史的に、FDA の規制上の決定 (製品ラベルにどの情報を含めるかなど) は先例に基づいており、Covid mRNA ショットは FDA の歴史的および法的先例からの明らかな逸脱でした。注目すべきデータの欠如と明確さの欠如は、ある種の時代を思い出させます。 モーリーの肝臓と腎臓のコーディアル 1800年代後半。違いは、当時、FDA は存在しませんでしたが、現在は約 20,000 人の職員を擁する FDA があり、少なくともそのうちの何人かは表向き、このラベルが透明で「真実」であると信じていたということです。
誰も正確に判断できない未知の/解読不可能な/あいまいな成分を記載することは、おそらく 1906 年の純粋食品医薬品法議員が「真実の表示」に関する FDA 規則を定めたときに意図したものではありません。それとは別に、異なるメーカーでは容量ごとに用量が XNUMX 倍になっているという事実 (30mcg/0.3mL vs 100mcg/0.5mL) は、これらの mRNA 配列のヌクレオチド長が大きく異なっているように見えることを意味します。 より多くの異なる LNP とアタッチメント。それは、スパイクタンパク質の転写に使用される mRNA 配列のサイズが、異なるメーカーと比較して約 10 倍であることを意味しますか (0.1 mcg/20 mL 対 0.1 mcg/XNUMX mL)、それともヌクレオチドの長さの違いに他の何かが寄与しているのでしょうか?
まだここまで読んでいる一般人のために(ちなみに、称賛):詳細なラベル情報の欠如は、売りに出されている住宅を大々的に宣伝し、セメント板の上に木とレンガでできていると宣伝しているのと同じかもしれないが、表示されていない家の写真(シーケンスなど)、面積を共有していないもの(分子量など)。いずれにせよ、情報不足は不十分であり、従来の基準から逸脱しています。
他のすべての FDA 承認薬(他の mRNA 薬を含む)には、製品に関する全成分開示が含まれています。 構造表現と分子量 人々が自分が何を得るのかを正確に知ることができるようにするためです。
それは本当です。思いつく限りの薬物を調べてください。 Drugs.com データベース すべてのラベルが構造および/または分子量をどのように提供するかに注目してください。 Covid mRNAショットが 目立つ これは、歴史的な FDA 承認慣行と「真実のラベル」規則の例外です。
2023年のデンマークの研究は、mRNA Covid-19 mRNA注射のバッチ間の重大な臨床的ばらつきを詳細に示しています:
潜在的に無効な「郵送」検査の検証さえも透明性を持たないことで、製造業者はFDAが監督するもう一つの非常に重要な部分、つまりmRNAショットのロット/バッチの変動による潜在的な臨床症状を見逃してしまったようだ。回顧展 デンマークの安全性研究 2023年初めに発表された論文では、ファイザー・バイオNテック社のBNT162b2 mRNA注射による有害事象報告の非常に逸脱したパターンを、デンマークのDKMA有害事象報告システムと相関させて詳述した。
次の折れ線グラフでは、異なる色の点がファイザー-BioNTech の mRNA 注射の異なるバッチを表しています。バッチを XNUMX つの異なるカテゴリに分類しました。高-低-(ほぼ)存在しない、報告された有害事象グループの数(それぞれ青、緑、黄色のプロット)。
言い換えると: 同じメーカーの推定上の「同等」製品は、バッチごとに有害事象の発生率が大きく異なるようで、各バッチは数十万回の mRNA 注射に相当します。
対応する線形回帰直線を追加すると、特定のパターンが現れました。
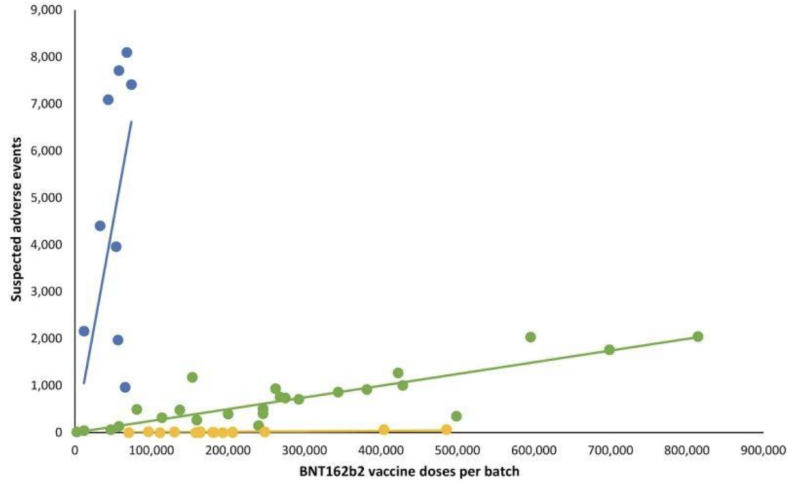
Covid-19 mRNA バッチ間の注目すべき有害事象の差異に関する重要な疑問は次のとおりです。
- 有害事象の差異は、バッチ間の mRNA 配列または mRNA 鎖の数の定性的または量的差異によるものでしょうか?
- 有害事象の差異は、バッチ間の LNP のサイズ/形態または量の定性的または定量的な差異によるものでしょうか?どのようなテストが行われたか 各種LNPの安全性を確保 mRNA注射に使用されますか?
- 黄色、緑色、青色のデータ ポイントに対応するバッチは、何らかの形で質的または量的に異なっていましたか?
- 管理施設 (またはサプライチェーンの他の場所) での製造後の保管/取り扱いが損なわれ、製品のばらつきが生じていましたか?
- 特定の製造施設/製造を担当する当直長に由来するこの製品および他の製品のシグマ/エラー率はどれくらいですか?
- これらのCovid mRNA製品の成分は、バッチに応じてインドまたは中国から調達されたものでしょうか、それとも他の国から調達されたものでしょうか?
- 最初から現在まで、Covid mRNA 製品のバッチのうち、FDA 検査官による対面採取によって検査されたのと、「郵送」されて検査されたのは何パーセントですか?すべてのバッチは、これら XNUMX つの収集方法のいずれかのみを使用してテストされましたか?
- FDA はデンマークの DKMA 有害事象報告システムのロットに対して放出試験検証を実施しましたか?もしそうなら、なぜ FDA はその特定の検査結果を公表しないのでしょうか?そうでない場合、なぜテストが行われなかったのでしょうか?
- LNP および/または mRNA 配列を汚染なく確実に一貫して生成することに関して根本的な問題はありますか?
デンマークの研究の結果と有害事象に関する上記の疑問は「対処し始める」ことができますが、FDA が放出試験の結果を独自に共有することがなければ解決しません。現状では、FDA (b)(4) の編集が遍在しているため、Covid mRNA ショットをテストするための検証された方法論を誰も知りません。 or デンマークの研究でどのロットがテストされたのか、またはテストされなかったのかを正確に示す or これらのバッチテストの結果。
…では、たとえ FDA がそれらのバッチテスト結果を公表することを選択したとしても、どのサンプルを「郵送」するかはメーカーが自ら選択しているため、消費者はそれらの結果が指定されたバッチを代表するものであるかどうかをどうやって知ることができるのでしょうか?
成分の透明性を提供せず、適切なサンプリング方法によって品質を保証することは、FDA の基本的かつ基本的な要件です。実際、それが FDA 設立の主な理由でした。米国人は、我が国の医薬品に関して、特にこれらの法律が100年以上前に制定されたものであるため、より透明性、監視、そして「真実の表示」に関する法律を強化されるべきではないでしょうか?
の下で公開 Creative Commons Attribution4.0国際ライセンス
再版の場合は正規リンクをオリジナルに戻してください。 褐色砂岩研究所 記事と著者。








