
新型コロナウイルスのパンデミック中、米国政府は数億人を保護、診断、治療することを目的とした約400の製品に数十億ドルを費やしたが、そのすべてに「EUA」または「緊急使用許可」のラベルが付いていた。
しかし、EUA とは実際には何を意味するのでしょうか?
その質問に答える前に、医療製品を認可または承認するための他の経路との関係で EUA がどのような立場にあるのかを理解するために、次のことを確認することが役に立ちます。 EUA ではないもの:
EUA は臨床試験中の実験製品の指定ではありません
EUA について 1 つだけ理解するとしたら、これは次のとおりです。EUA は、FDA 規制またはその他の法的要件によって管理される臨床試験を受けている製品には適用されません。
また、EUA は、重度の難病患者に完全に承認される前に実験製品へのアクセスを許可するために適用される、「思いやりのある使用」アクセスと呼ばれることが多い拡張アクセス使用 (EAU) とも同じではありません。
このテーブルは、 FDA-CDC 2020 プレゼンテーション 臨床試験中の製品、拡大された「思いやりのある」アクセスを通じて患者に提供される製品、および EUA を通じて認可された製品の違いを要約します。
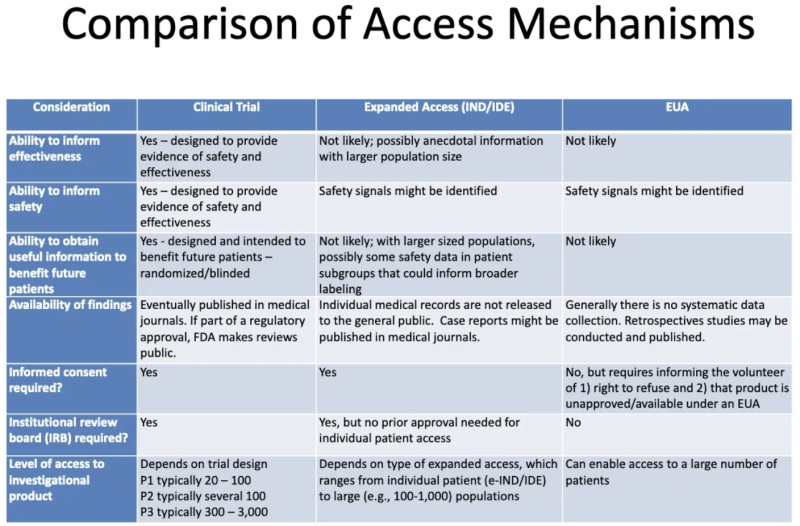
この表から EUA についてわかることは次のとおりです。
- EUA を付与するプロセスでは、製品の有効性に関する情報が生成される可能性はほとんどありません。
- EUA を付与するプロセスは、安全性または有効性の証拠を提供するように設計されていませんが、安全性の兆候が特定される可能性があります。
- 製品が EUA を取得して一部の患者に投与されると、将来の患者に利益をもたらす有用な情報が得られる可能性はほとんどありません。
- EUA には有効性や安全性に関する体系的なデータ収集がなく、規制当局の承認プロセスの一環として医学雑誌にデータが掲載されることもありません。
- インフォームド・コンセントは必要ありませんが、製品の摂取を「自発的に」行う患者には、拒否できること、およびその製品がEUAの下で未承認/入手可能であることを伝えなければなりません。
- 治験審査委員会(IRB)は必要ありません。 [IRB 臨床試験において被験者の健康を保護することを目的とした委員会です]
EUA が通常の承認プロセスからどのように分離されているかをさらに明確にするために、 2009 年国立アカデミー医学研究所の出版物、次のステートメントが見つかります。
EUA は開発経路の一部ではないことを認識することが重要です。これは緊急事態時にのみ使用される完全に別個の組織であり、医薬品承認プロセスの一部ではありません。 (p.28)
要約すると:
製品の EUA を付与するプロセスでは、安全性または有効性の証拠が生成される可能性はほとんどありません。製品が EUA を取得して患者に投与されると、有効性や安全性に関する体系的なデータ収集が存在しないため、将来の患者に利益をもたらす有用な情報が得られる可能性はほとんどありません。
CDC/FDA および IMNA からのこれらすべての非常に明確な情報に基づいて、緊急使用許可は、非常に慎重に、悲惨な緊急事態の場合にのみ適用されるべきプロセスであると結論付けるのは公平でしょう。
次に、EUA が法的にどのような種類の緊急事態に対処するように設計されているかを見てみましょう。
EAU は大量破壊兵器緊急事態を対象としています
上記の EUA の「アクセスメカニズム」を許可する法律は、CBRN (化学、生物、放射線、核) 物質とも呼ばれる大量破壊兵器 (WMD) が関与する極端な差し迫った緊急事態の場合のために作成されました。
食品医薬品局のやり方はこうだ (FDA) EUA の権限について説明:
FD&C 法第 564 条 (21 USC 360bbb–3)により、FDA は生物剤、化学剤、核剤、放射性物質に対する公衆衛生保護を強化することができます。
この EUA 権限により、FDA は、生物学的、化学的、核的、または放射性物質によって引き起こされる重篤または生命を脅かす疾患や状態を診断、治療、または予防するために、適切で承認された適切な医薬品がない場合に、緊急時に医療対策を確実に使用できるように支援することができます。 、および利用可能な代替案(他の基準とともに)。
これらの EUA 権限は、CBRN エージェントによる攻撃への備えに関連する非常に特殊な状況下で 2004 年に付与されました。
説明したように ハーバード大学法の健康法案に記載されている,
結局のところ、緊急使用許可を生み出すのは対テロ戦争だった。 11 年 2001 月 XNUMX 日の事件とその後の炭疽菌郵便攻撃の後、議会は 2004 年プロジェクト バイオシールド法.
記録 これは、議会が自然発生的なパンデミックへの備えではなく、特にバイオテロの脅威に焦点を当てていたことを示している。
大量破壊兵器攻撃を伴うこのような狭い種類の真に極端な緊急事態を考えると、EUA の「アクセスメカニズム」が多くの規制監視や製造基準や臨床試験基準の遵守を必要としない理由は理解できます。
では、EUA アクセス メカニズムには実際に何が必要なのでしょうか?
緊急使用許可 (EUA) の 3 つのステップ
医療製品に EUA が付与されるには、次の 3 つのことが起こる必要があります。
- 国土安全保障長官、国防長官、保健福祉長官は、CBRN エージェントによる攻撃または攻撃の脅威、またはそのようなエージェントによって引き起こされる病気を伴う緊急事態が発生していると判断する必要があります。
- FDAはEUAを発行する際に4つの「法定基準」を満たしていることを確認する必要がある。
- FDAはEUAに「一定の必須条件を課す」必要がある。
EUA ステップ 1: CBRN 緊急事態の宣言
EUA の緊急事態宣言は別個のものであり、大統領、HHS 長官、その他の者が発令する可能性のある他の緊急事態宣言とは無関係です。これは特に EUA を発動する目的で発令される必要があり、他の緊急事態宣言とは無関係に終了または延長することができます。
ここに EUA法には次のように書かれています EUA の「アクセス メカニズム」を有効にするために考えられる 4 つのシナリオは次のとおりです。
- 生物剤、化学剤、放射線剤、または核剤による攻撃の危険性が高まる国内緊急事態が存在する、または国内緊急事態の重大な可能性があるとの国土安全保障長官の判断。
- 軍事的緊急事態が存在する、または軍事的緊急事態が発生する重大な可能性があり、危険の増大を伴うと国防長官が判断したこと。 ユナイテッド 米国 タイトル 10 またはタイトル 50 の権限の下で活動する人員を含む軍隊は、次のものによる攻撃を行う。
- 生物学的、化学的、放射性物質、または核物質、または複数の物質。または
- ユナイテッド航空に差し迫った生命を脅かす特定のリスクを引き起こす可能性がある、または関連する単数または複数の薬剤 米国 軍事力;
- による決定 秘書 [保健福祉サービスの]国家安全保障または国民の健康と安全に影響を与える、または影響を与える重大な可能性がある公衆衛生上の緊急事態、または公衆衛生上の緊急事態が発生する重大な可能性があること。 ユナイテッド 米国 海外に居住しており、生物学的、化学的、放射性物質、または核物質、またはそのような物質に起因する可能性のある病気や状態に関係する国民。または
- のセクション 319F–2 に基づく重大な脅威の特定 公衆衛生局法 [42 USC 247d–6b] 国家安全保障または国民の健康と安全に影響を与えるのに十分な ユナイテッド 米国 海外に住む国民。
EUA ステップ 2. 法定基準を満たす
長官の一人がEUAを正当化する緊急事態であると宣言すると、FDAがEUAを発行するにはさらに4つの「法定基準」を満たす必要がある。 FDA はこれらの要件を次のように説明しています:
- 重篤または生命を脅かす病気または状態
FDA が EUA を発行するには、HHS 長官の EUA 宣言で言及されている CBRN 薬剤が、重篤または生命を脅かす疾患または症状を引き起こす可能性がある必要があります。
- 有効性の証拠
EUA の対象として検討される可能性のある医療製品は、HHS 長官の宣言で特定された CBRN 病原体によって引き起こされる可能性のある重篤または生命を脅かす疾患や症状の予防、診断、または治療に「効果がある可能性がある」製品です。第 564 条 (b) に基づく緊急事態または緊急事態の脅威。
EUA の「有効である可能性がある」基準は、FDA が製品承認に使用する「有効性」基準よりも低いレベルの証拠を提供します。 FDA は、以下で説明するように、リスク利益分析を使用して、可能性のある EUA 製品の潜在的な有効性をケースバイケースで評価する予定です。
[太字を追加]
- リスクと利益の分析
特定の疾患または症状の診断、予防、または治療に使用した場合の製品の既知および潜在的な利点が、製品の既知および潜在的なリスクを上回ると長官が判断した場合、製品は EUA の対象とみなされる可能性があります。
製品の既知および潜在的な利点が既知および潜在的なリスクを上回るかどうかを判断する際に、FDA は 見るつもりです 総合的なリスクと利益の判断を行うための科学的証拠を総合的に判断します。そのような証拠は、 起こるかもしれない さまざまな情報源から、 含めることができます (ただしこれらに限定されません):国内外の臨床試験結果、動物モデルによるin vivo有効性データ、in vitroデータ、 FDA の検討に利用可能。 FDA はまた、製品の質と量も評価します。 入手可能な証拠、現在の科学的知識を考慮すると。
[太字を追加]
- 代替手段なし
FDA が EUA を発行するには、疾患または状態の診断、予防、または治療のための候補製品に代わる適切で承認済みの利用可能な代替品が存在していなければなりません。緊急ニーズを完全に満たすために承認された代替品の供給が不十分な場合、潜在的な代替製品は「利用できない」とみなされる場合があります。
EUA ステップ 3. 必要な条件を課す
EUA 固有の緊急事態宣言が発令され、FDA がその製品が有効である可能性があり、利用可能な証拠が何であれその利点がリスクを上回ると判断すると、関連する規制がさらに 1 層追加されます。
方法は次のとおりです。 EUAに関する2018年議会調査局報告書 これを説明します:
FFDCA §564 は、EUA に特定の必須条件を課すよう FDA に指示し、必要に応じて追加の裁量条件を許可します。必要な条件は、EUA が未承認製品に対するものであるか、承認済み製品の未承認使用に対するものであるかによって異なります。未承認の製品の使用条件は次のとおりです。
(1) 製品を投与する医療専門家が必要な情報を確実に受け取れるようにする。
(2) 製品が投与される個人が必要な情報を確実に受け取れるようにする。
(3) 製品に関連する有害事象の監視と報告を提供する。そして
(4) 製造業者による記録保持と報告を提供する。
まとめ
この記事で述べたように、FDA/CDC は、緊急使用許可 (EUA) を付与するプロセスでは製品の有効性や安全性に関する情報が生成される可能性は低いことを明確に認識しています。 EUA を管理する法律の条文を見ると、これが確かに正しい評価であることがわかります。
EUA 法は、製品の安全性や有効性を決定する可能性のある法的基準や規制基準を課すものではありません。唯一の基準は、その製品が有効である可能性があり、既知の利点が既知の害を上回るとFDAが信じているかどうかです。製品が医薬品承認プロセスを経ていないため、既知の害や既知の利点がない場合、FDA はその決定を行うために選択した情報や基準を使用できます。
これらすべてのことから、製品が EUA の候補である企業は、選択したあらゆる手段を通じて製品の安全性および/または有効性を実証しようとする可能性があるということになります。そのような試みの有無(臨床試験であれ、その他のデータ収集メカニズムであれ)、およびその試みがどのように実施されるかは、すべて企業次第です。 EUA 法のいかなる規定も、企業が追求することを選択した研究やその他のデータ収集メカニズムを設計、実施、または分析する方法には適用されません。
これは、Covid 製品に適用されると次のことを意味します。
- 新型コロナウイルス製品が EUA を取得するためには、臨床試験からの安全性や有効性のデータは必要ありませんでした。
- EUA プロセスで参照される臨床試験はすべて、法的に適用される規制基準を無視して実施されました。
- これらの製品に有効性や安全性が欠けていることがわかっても、それは驚くべきことではありません。それはプロセスの結果である可能性が非常に高いです。
- 製品の安全性または有効性に関する非 EUA 決定の基礎となる EUA プロセスからのデータはありません。したがって、EUA 以外で製品を使用するには、最初から通常の医療製品の法的承認プロセスを通過する必要があります。
新型コロナウイルスワクチンの承認プロセスの詳細 こちら.
著者からの転載 サブスタック
の下で公開 Creative Commons Attribution4.0国際ライセンス
再版の場合は正規リンクをオリジナルに戻してください。 褐色砂岩研究所 記事と著者。








